Для перегляду доступних досліджень та термінів їх виконання








Загальний варіабельний імунодефіцит: причини, дебют, клінічні прояви, діагностика та лікування
09.02.2023
Загальний варіабельний імунодефіцит (ЗВІД, Common variable immunodeficiency, CVID) – група первинних імунних розладів, що належать до вроджених помилок імунітету з різними механізмами розвитку, що характеризується критично низьким рівнем продукції антитіл і, відповідно, порушенням імунного захисту. Термін «загальний» є перекладом англійського слова «common», яке насправді означає «частий». ЗВІД дійсно належить до найбільш поширених важких первинних імунодефіцитів. Слова «варіабельний» відображає різноманіття клінічної маніфестації. ЗВІД є хронічним невиліковним захворюванням тривалістю в життя, але ефективні засоби підтримуючої терапії дозволяють особам зі ЗВІД вести майже нормальне продуктивне життя. Саме тому вчасне розпізнавання симптомів і правильна діагностика є надзвичайно важливими для порятунку життя пацієнтів і забезпечення його високої якості.
Частота захворювання оцінюється як 1 на 25-50 тисяч населення. ЗВІД належить до вроджених імунодефіцитів з типовим дебютом в дорослому віці. Захворювання також відоме як гіпогаммаглобулінемія, агаммаглобулінемія дорослого віку, гіпогаммаглобулінемія з пізнім початком, набута агаммаглобулінемія. Незважаючи на те, що діагноз зазвичай припадає на вік 20-50 років (згідно з дослідженням Європейського реєстру імунодефіцитів середній вік появи симптомів – 26,3 роки), перші ознаки можуть з’явитися у дитячому або підлітковому віці, що часто залишаються нерозпізнаними. Жінки й чоловіки хворіють однаково часто.
Причини
Більшість випадків є спорадичними і виникають у людей без обтяженого сімейного анамнезу. Найбільш ймовірно, захворювання є результатом взаємодії поламок в генах, що відповідають за розвиток і функціонування імунної системи, і зовнішніх факторів. Генетичну природу ЗВІД вдається встановити лише у 10-15% випадків. Мутації щонайменше в 13 генах були асоційовані з розвитком загального варіабельного імунодефіциту: TNFRSF13В, CD19, CD81, CR2, ICOS, IKZF1, IL21, LRBA, MS4A1, NFKB1, NFKB2, PRKCD, TNFRSF13C.
Деякі з цих мутацій успадковують за аутосомно-рецесивним типом, інші – аутосомно-домінантним.
Найбільш часто ідентифікованими є мутації в гені TNFRSF13B, часто ці мутації є спорадичними, які виникають під час дозрівання статевих клітин або в ранньому ембріональному періоді. Мутації в гені TNFRSF13B можуть успадковуватися як за аутосомно-рецесивним, так і аутосомно-домінантним типом. Білок, що є результатом роботи даного гену, відіграє роль у виживанні й дозріванні В-лімфоцитів і в продукції антитіл. Інші гени, асоційовані зі ЗВІД, також залучені у функціонування і/або дозрівання імунних клітин. В багатьох пацієнтів зі ЗВІД виявляють поліморфізм в гені TACI. Тим не менше, ця генетична аномалія лише підтверджує ризик розвитку ЗВІД, але сама по собі не є його єдиною причиною, оскільки не в усіх членів родини з однаковою мутацією розвивається захворювання. Додаткові неідентифіковані на сьогодні тригери, очевидно, необхідні для початку захворювання. Подальші дослідження для ідентифікації молекулярно-генетичних причин розвитку ЗВІД на сьогодні активно продовжуються, щороку приводячи до відкриття нових мутацій і генів.
Таким чином, ЗВІД - це гетерогенна група імунодефіцитних станів з різними механізмами успадкування, пусковими факторами і генетичними поламками, що лежать в їх основі.
Тим не менше, спільними для них є порушення антитілопродукції. В усіх осіб із загальним варіабельним імунодефіцитом спостерігається значне зниження продукції щонайменше двох класів імуноглобулінів (IgG та IgA) або більше. У половини пацієнтів спостерігається також майже нульовий рівень продукції імуноглобуліну М та інших класів. Вірогідно, що імунологічні порушення розвиваються поступово. Вважають, що ЗВІД пов’язаний із селективним дефіцитом імуноглобуліну А. В більшості випадків селективний дефіцит імуноглобуліну А є самостійним захворюванням, ізольоване порушення вироблення імуноглобуліну А може передувати розвитку розгорнутої картини ЗВІД, що підтверджується наявністю випадків існування в одній родині осіб із селективним дефіцитом імуноглобуліну А та ЗВІД.
Клінічні прояви
Різноманіття перших клінічних проявів і їх важкість суттєво варіюють у різних пацієнтів.
Основним клінічним наслідком гіпогаммаглобулінемії є розвиток рецидивних бактеріальних інфекцій. До додаткових порушень при ЗВІД належать підвищений ризик розвитку аутоімунних, автозапальних, алергічних, гастроінтестинальних, лімфопроліферативних та онкологічних захворювань.
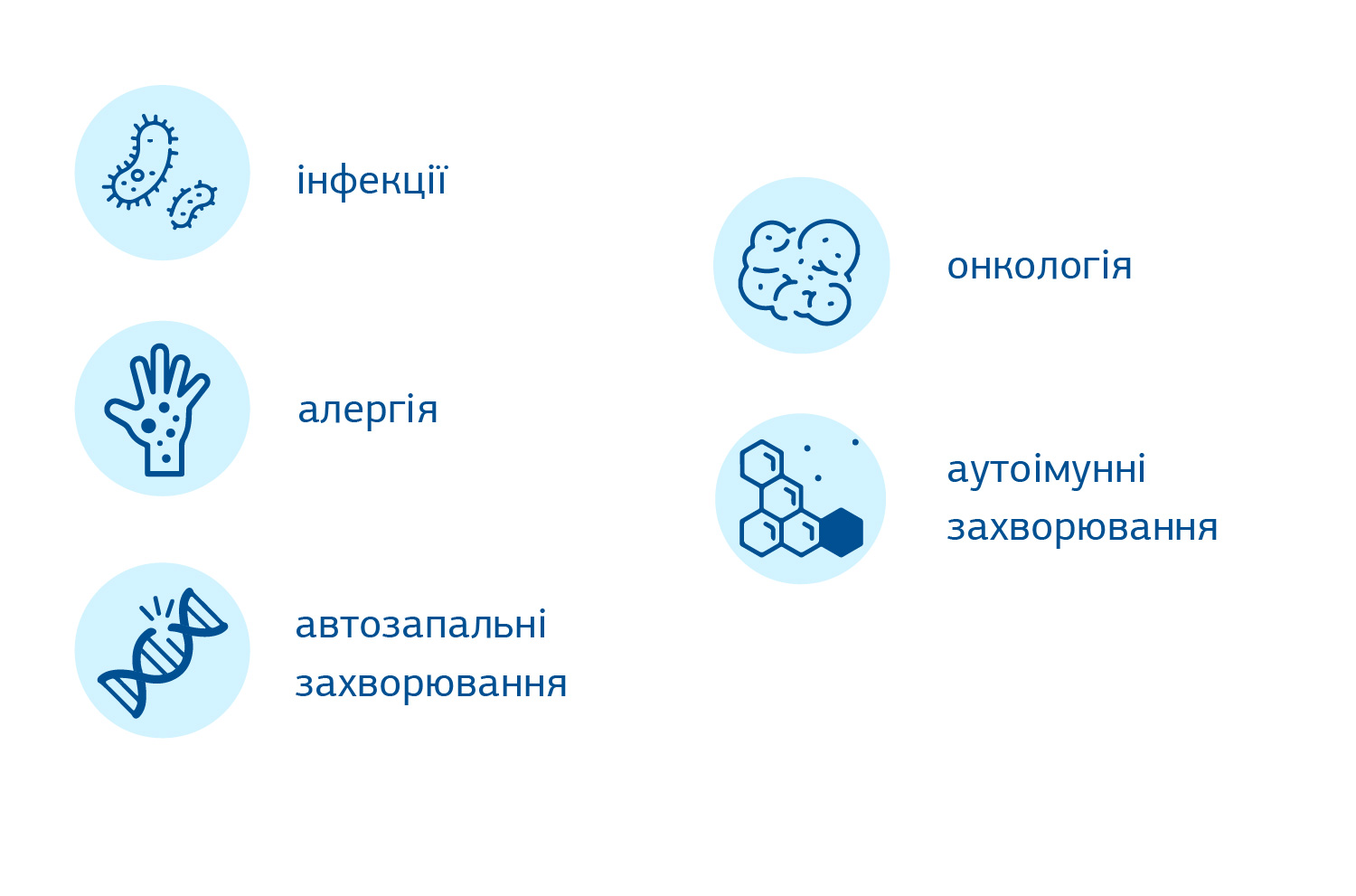
Інфекції є прямим наслідком низького (в ряді випадків – близького до нульового) рівня циркулюючих антитіл, що не здатні захистити проти мікробних агентів. Найбільш частими мікробами, які викликають захворювання у пацієнтів зі ЗВІД, є Haemophilus influenzae, Streptococcus pneumoniae і Staphylococcus aureus, рідше - Neisseria meningitidis, Pseudomonas aeruginosa і Giardia lamblia. Найбільш частими проявами є інфекції дихальних шляхів (носоглотка, приносові пазух, бронхи, легені) і вух, але фактично можуть охоплювати будь-які органи, включаючи шкіру і підшкірну клітковину, шлунково-кишковий тракт. Зазвичай інфекції добре відповідають на лікування антибіотиками, проте швидко рецидивують після їх відміни. Безперервно рецидивуючі бронхо-легеневі інфекції можуть спричиняти перманетне ушкодження бронхів – бронхоектази.
Пацієнти зі ЗВІД також знаходяться в ризику розвитку вакцин-контрольованих інфекцій, таких як кір, грип, дифтерія, правець та інші, оскільки імунний захист від раніше отриманих щеплень втрачається.
Порушення захисних механізмів негативно впливають не тільки на контроль над інфекціями, а й на механізми імунологічної толерантності. Від 20 до 50% пацієнтів зі ЗВІД мають аутоімунні розлади, такі як аутоімунні цитопенії (гемолітична анемія, тромбоцитопенія, аутоімунна нейтропенія), ревматоїдний артрит, ЮІА, васкуліт, СЧВ, гломерулонефрит, аутоімунний гепатит, целіакія, атрофічний гастрит і перніціозна анемія, ендокринопатії (аутоімунний тиреоїдит, цукровий діабет 1 типу, Аддісонова хвороба або гіпопаратиреоїдизм), псоріаз, алопеція, вітиліго. Найбільш часто аутоімунним атакам піддаються клітини крові, з яких тромбоцитопенія посідає перше місце і нерідко є першою маніфестацією цього захворювання, ще до розвитку інфекційного синдрому. Аутоантитіла, первинна або вторинна дисфункція кісткового мозку (КМ), клітинний аутоімунітет і дизрегуляція імунної системи є основними причинами цих проявів. Водночас як і у випадку патологічної схильності до інфекційних захворювань частіше підозрюють вроджений імунодефіцит, перевірка пацієнтів із аутоімунними захворюваннями не є частою практикою, що затримує шлях до правильного діагнозу.
Типовими для пацієнтів із ЗВІД є хронічна діарея, синдром мальабсорбції і втрата маси тіла. Стійкий до лікування перебіг хронічної діареї може бути важливою вказівкою на наявність імунодефіциту. Симптоми ентеропатії при ЗВІД можуть бути наслідком як інфекцій (наприклад, персистенції Giardia lamblia, кампілобактерами, ЦМВ або сальмонел), так і запального захворювання кишечника, що вимагає проведення диференційної діагностики між ними.
Через порушення розвитку В-лімфоцитів можлива акумуляція їх в лімфоїдних тканинах з розвитком лімфопроліферативного синдрому: збільшення лімфатичних вузлів, гепато- та спленомегалія, формуванням гранульом в різних органах (утворення третинної лімфатичної тканини, зокрема в легенях та шлунково-кишковому тракті). Helen Chapel із співавторами (2008) у своїй групі 334 пацієнтів із ЗВІД з 7 європейських центрів виявили ознаки лімфопроліферації у 54 % хворих.
В багатьох пацієнтів із загальним варіабельним імунодефіцитом мають в анамнезі симптоми, схожі на алергію. В той же час симптоми рецидивних респіраторних інфекцій можуть бути сплутані з алергією, виступати їх маскою. За даними літератури, астма та алергічний риніт зустрічаються у 37,5% та 55,5% пацієнтів із ЗВІД, відповідно. Тим не менше, оскільки алергія є дуже поширеним захворюванням, особливо в дитячому віці, і ці показники постійно зростають у всьому світі, такий прояв є показовим переважно тільки в комбінації з іншими, більш специфічними проявами.
Тривале порушення функції імунітету з часом призводить до підвищеного ризику онкологічних захворювань. У випадку із ЗВІД є особливо схильність до розвитку лімфом і раку шлунку, ризик розвитку даних видів раку в 50 разів перевищує ризик в загальній популяції. Іноді захворювання може маніфестувати з лімфоми.
Імунологічна характеристика
У пацієнтів із ЗВІД спостерігається різноманіття імунологічних фенотипів. Зокрема, широко варіюють рівні сироваткових імуноглобулінів. Пацієнти можуть бути згруповані в наступні три категорії: відсутня продукція імуноглобулінів; частково збережена продукція імуноглобулінів; збережена продукція лише імуноглобуліну М.
Кількість В-лімфоцитів також значно варіює у різних пацієнтів. У близько 12% пацієнтів В-лімфоцитів повністю відсутні, але 54% мають рівень В-лімфоцитів в межах референтних значень. Особливістю ЗВІД є зниження відсотку В-клітин пам’яті. Також може відмічатися порушення відносної і абсолютної кількості інших субпопуляцій В-лімфоцитів. В деяких пацієнтів відмічається також варіабельне порушення Т-клітинної ланки, зокрема, часто відмічається низький вміст CD4+ і Т-регуляторних клітин. Близько 10% можуть мати рівень CD4+ T-клітин на рівні нижче, ніж 200 клітин/мм3; такий фенотип ще називають «комбінований імунодефіцит із пізнім початком», цей різновид захворювання має гірший прогноз, ніж класичний ЗВІД.
Діагностика
Через те, що в більшості випадків генетичний дефект встановити не вдається, на сьогодні діагноз загального варіабельного імунодефіциту базується на клініко-лабораторних критеріях. Для встановлення клінічного діагнозу без генетичного діагнозу Європейським товариством імунодефіцитів (ESID) розроблені робочі діагностичні критерії.
Робочі діагностичні критерії ESID-2019:
принаймні одне з наступного:
- підвищена сприйнятливість до інфекції
- аутоімунні прояви
- гранулематозна хвороба
- поліклональна лімфопроліферація неясного генезу
- сімейний анамнез дефіциту антитілоутворення
ТА - зниження IgG (<4,5 г/л) і суттєве зниження рівня IgA з або без зниження IgM (виміряні щонайменше двічі; <2SD від нормальний рівень для їхнього віку)
ТА
принаймні одне із наступного: - знижена продукція антитіл на вакцини і/або відсутні ізогемаглютиніни;
- знижені В-клітини пам’яті
ТА - вторинні причини гіпогамаглобулінемії виключено.
Загальний варіабельний імунодефіцит встановлюється у осіб старше 4 років згідно з перерахованими критеріями, якщо альтернативні причини гіпогаммаглобулінемії, такі вторинні гіпогаммаглобулінемії і інші відомі первинні імунодефіцити виключені.
Лікування
Основою лікування ЗВІД є замісна терапія імуноглобулінами. Регулярне введення імуноглобулінів внутрішньовенним (1 раз на 4 тижні) або підшкірним (1-3 рази на тиждень) шляхом в більшості випадків компенсує дефект антитілоутворення і дозволяє вести хворим практично нормальне життя. Інколи початок замісної терапії значно полегшує перебіг навіть неінфекційних ускладнень, таких як артрити, шкірні ураження, алергічні прояви. У пацієнтів із частково збереженою функцією антитілопродукції може застосовуватись додаткова вакцинація проти пневмококової і менінгококової інфекцій та грипу. При наявності хронічних вогнищ бактеріального запалення, особливо у пацієнтів із сформованими бронхоектазами, на додаток до замісної терапії призначається тривала антибіотикопрофілактика. Лікування інших ускладнень, таких як ураження шлунково-кишкового тракту, аутоімунні, алергічні чи онкологічні захворювання у разі виникнення здійснюється згідно з протоколами лікування даних захворювань.
Клінічний приклад:
Чоловік 36 років звернувся з приводу безперервно рецидивуючого гнійного синуситу і частих епіходів гнійного отиту протягом останніх двох років, що призвело до часткової втрати слуху. В анамнезі – перенесена у віці 33 років неходжкінська В-клітинна лімфома, з приводу якої пацієнт отримував ритуксимаб, На фоні лікування була виявлена гіпогаммаглобулінемія (IgA 0,15 г/л, IgM 2,5 г/л, IgG 1,8 г/л), розцінена як побічний ефект терапії. На момент звернення, через 3,5 роки ремісії і відміни ритуксимабу в пацієнти визначаються нормальні рівні всіх субпопуляцій лімфоцитів, включаючи В-лімфоцити і рівні всіх імуноглобулінів – нижче межі визначення.
Тривалий час, що минув після відміни ритуксимабу, так само як і нормальна кількість загальних В-лімфоцитів, що відновились після застосування моноклональних антитіл проти них, робить сумнівним трактування гіпогаммаглобулінемії як наслідку терапії ритуксимабом.
При підозрі на загальний варібаельний імунодефіцит, який маніфестував з лімфоми для перевірки відповідності критеріям були проведені додаткові обстеження:
Ізогемаглютиніни α, β (природні антитіла за системою ABО)
– дозволяє оцінити здатність до спонтанної продукції природніх імуноглобулінів класу М і здатності до продукції антитіл проти полісахаридних антигенів (один із необхідних критеріїв)
В-клітинна ланка (В1-, В2-, В-клітини пам'яті) – дозволяє оцінити рівень В-клітин памяті (один з необхідних критеріїв)
За результатами обстеження:
при групі крові А(ІІ) в пацієнта бета-ізогемаглютиніни не виявлені (референтні значення 1:8-1:128)
В-клітини пам’яті – 6,09% (референтні значення 22-40%).
Таким чином, клініко-лабораторні критерії дають право встановити діагноз загального варіабельного імунодефіциту.
В цьому випадку лімфома могла розвинутись на фоні вже порушеного імунітету як перший його прояв (відсутність даних попереднього імунологічного обстеження робить неможливим встановлення причинно-наслідкового зв’язку) або можливо, лімфома або її причина виступила як тригер запуску генетичного дефекту у схильного до цього пацієнта.
Даний випадок демонструє складність іноді диференційної діагностики між первинним і вторинним імунодефіцитом, тому корисним було б мати інформацію про вихідний рівень імунологічних показників перед призначенням імуносупресивної терапії шляхом імунологічного обстеження (субпопуляції лімфоцитів і імуноглобуліни A, M, G). Також є ілюстрацією того, що випадки тривалого відновлення після імуносупресивної терапії – привід підозрювати первинний імунодефіцит із пізнім стартом, яким і є загальний варіабельний імунодефіцит.
Література:
- ПЕРВИННІ ІМУНОДЕФІЦИТИ КЛІНІЧНА НАСТАНОВА, ЗАСНОВАНА НА ДОКАЗАХ (2021) https://www.dec.gov.ua/wp-content/uploads/2022/01/2021_11_01_kn-pid.pdf
- Наказ МОЗ України № 2952 від 31.12.2021 Про затвердження Стандартів медичної допомоги "Діагностика та лікування первинних імунодефіцитів"
- Robert A Schwartz. Common Variable Immunodeficiency. https://emedicine.medscape.com/article/1051103-overview Updated: Jun 08, 2022
- Bonilla, Francisco A.; Geha/" rel="nofollow noopener noreferrer" target="_blank", Raif S. (2009). "Common Variable Immunodeficiency". Pediatric Research. 65 (5): 13R–19R. doi:10.1203/pdr.0b013e31819dbf88. PMID 19190529. S2CID 9361175.
- Kienzler AK, Hargreaves CE, Patel SY. The role of genomics in common variable immunodeficiency disorders. Clin Exp Immunol. 2017 Feb 25. [QxMD MEDLINE Link].
- Christiansen M, Offersen R, Jensen JMB, Petersen MS, Larsen CS, Mogensen TH. Identification of Novel Genetic Variants in CVID Patients With Autoimmunity, Autoinflammation, or Malignancy. Front Immunol. 2019. 10:3022.
- Agondi RC, Barros MT, Kokron CM, Cohon A, Oliveira AK, Kalil J, Giavina-Bianchi P. Can patients with common variable immunodeficiency have allergic rhinitis? Am J Rhinol Allergy. 2013 Mar-Apr;27(2):79-83. doi: 10.2500/ajra.2013.27.3855. PMID: 23562193.
- Schwartz, Robert A; Modak, Rohit; Modak, Prema. "Common Variable Immunodeficiency Treatment and Management". Medscape. Retrieved 17 February 2016.
- Робочі діагностичні критерії ESID-2019 https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria




